


nf-rnaSeqMetagen is a Nextflow
To use the nf-rnaSeqMetagen pipeline, the following are required:
- Software dependencies:
- RNA-seq data (paired-end for now - support for single-ended reads to follow)
- Reference genome (FASTA sequences) and its annotation file (GFT)
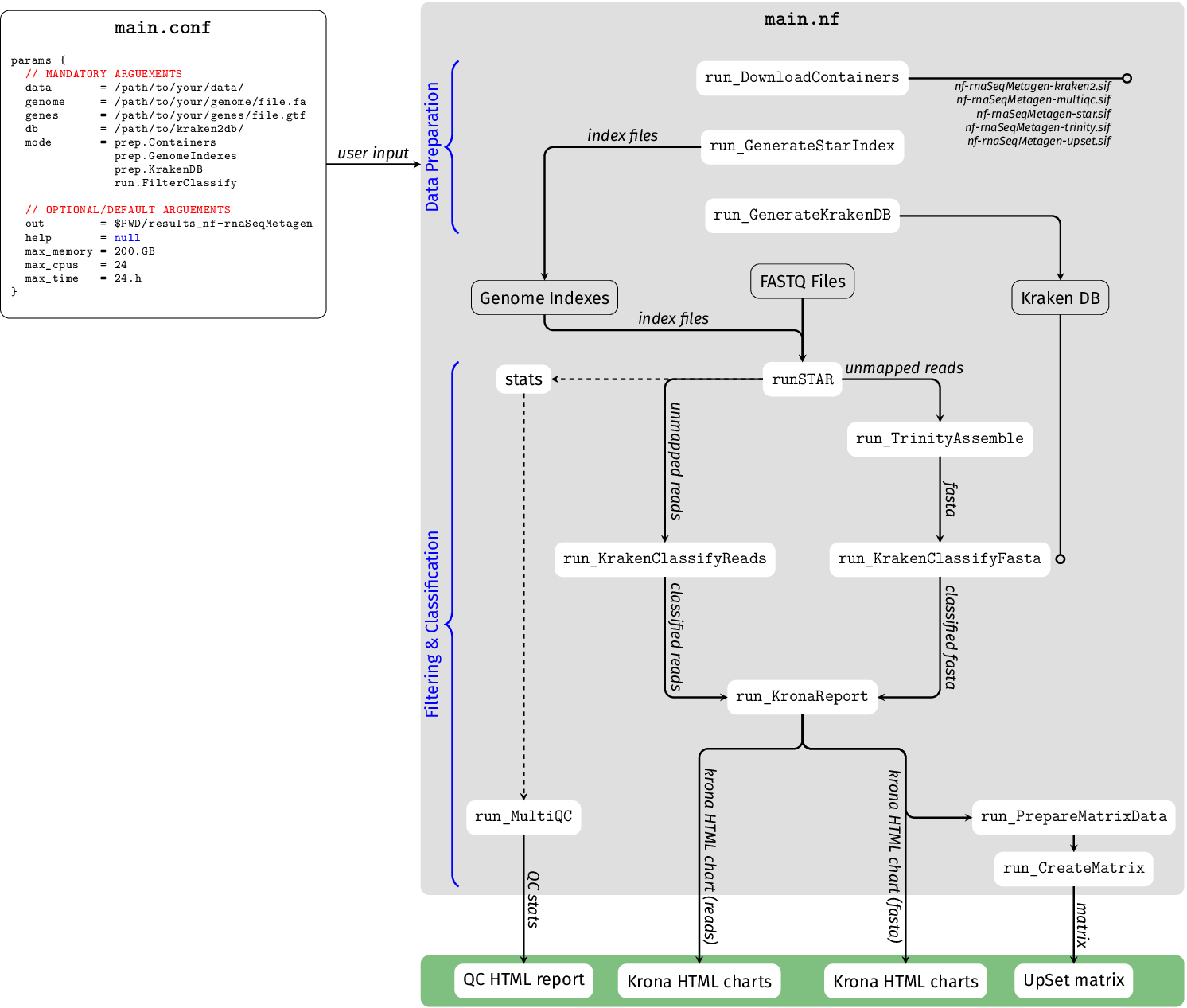
1. Obtaining the nf-rnaSeqMetagen Pipeline and Preparing Data
First, you need to clone the nf-rnaSeqMetagen repository onto you machine. You can either use git or nextflow. I recommend you use nextflow. The rest of this documentation assumes that you have used nextflow to clone this workflow.
nextflow pull https://github.com/phelelani/nf-rnaSeqMetagen
Content of the repository (located in $HOME/.nextflow/assets/phelelani/nf-rnaSeqCount):
To get the help menu for the workflow, execute the following command from anywherre on your system:
nextflow run nf-rnaSeqMetagen --help
1.1. Download test datasets (optional)
We will now download the reference genome (along with its annotation file) from Ensembl. We will also download the FASTQ files from the H3ABioNet site, which we will analyse using the nf-rnaSeqMetagen workflow. NB: Skip this section if you have your own data to analyse using this workflow! This section is only for getting data to practice using the nf-rnaSeqMetagen workflow!
Make directories:
mkdir example
cd example
mkdir reference
mkdir data
Download and decompress the mouse reference genome along with its annotation:
wget -c -O reference/genome.fa.gz ftp://ftp.ensembl.org/pub/release-68/fasta/mus_musculus/dna/Mus_musculus.GRCm38.68.dna.toplevel.fa.gz
wget -c -O reference/genes.gtf.gz ftp://ftp.ensembl.org/pub/release-68/gtf/mus_musculus/Mus_musculus.GRCm38.68.gtf.gz
gunzip reference/genome.fa.gz
gunzip reference/genes.gtf.gz
Download RNA-seq test dataset from H3ABioNet: script.
cd data
wget https://phelelani.github.io/nf-rnaSeqMetagen/examples/data/get_data.sh
sh get_data.sh
ls -l
cd ..
1.2. Download the singularity containers (required to execute the pipeline):
nextflow run nf-rnaSeqMetagen -profile slurm --mode prep.Containers
1.3. Generating genome indexes.
To generate the STAR genome indexes, run the following commands:
nextflow run nf-rnaSeqMetagen -profile slurm --mode prep.GenomeIndexes --genome "$PWD/reference/genome.fa" --genes "$PWD/reference/genes.gtf"
1.4. Creating the Kraken2 database:
To create the Kraken2 database, run the following command:
nextflow run nf-rnaSeqMetagen -profile slurm --mode prep.KrakenDB --db $PWD/K2DB
We are now ready to execute the workflow!
2. Executing the Main nf-rnaSeqMetagen Pipeline
As seen on the help menu above, there are a couple of options that you can use with this workflow. It can become a bit tedious and confusing having to specify these commands everytime you have to execute the each section for the analysis. To make your life easier, we will create a configuration script that we will use in this tutorial (we will pass this using the -c option of nextflow). You can name it whatever you want, but for now, lets call it myparams.config. We will add the mandatory arguements for now, but as you become more farmiliar with the workflow - you can experiment with other options. You can use your favourite text editor to create the myparams.config file. Copy and paste the the parameters below:
params {
data = $PWD/data
out = $PWD/myresults
genome = $PWD/reference/genome.fa
genes = $PWD/reference/gene.gtf
db = $PWD/K2DB
}
To perform filtering of host reads and classification of exogeneous reads, use this command:
nextflow run nf-rnaSeqMetagen -profile slurm --mode run.FilterClassify -c myparams.config
3. Exploring nf-rnaSeqMetagen Results
- [1] Sample analysis directories => `<output_directory>/<sample_1> .. <sample_N>`
- [2] MultiQC => `<output_directory>/MultiQC`
- [3] Upset tool => `<output_directory>/upset`
- [4] Workflow tracing => `<output_directory>/workflow-tracing
3.1. MultiQC
View the full MultiQC report here.
3.2. Sample analysis directories
3.2.1. Krona report: raw reads (SRR5074528)
View full Krona chart for raw reads here.
3.2.2 Krona report: assembled reads (SRR5074528)
View full Krona chart for assembled reads here.
3.3. UpSet visualisation tool
View full UpSet plot here.
3.4. Workflow tracing
3.4.1. Report
View full Nextflow report here.
3.4.2. Timeline
View full timeline report here.